SparseAEH: Scalable autoregressive expression histology discovery in spatial transcriptomics via sparse Gaussian kernels
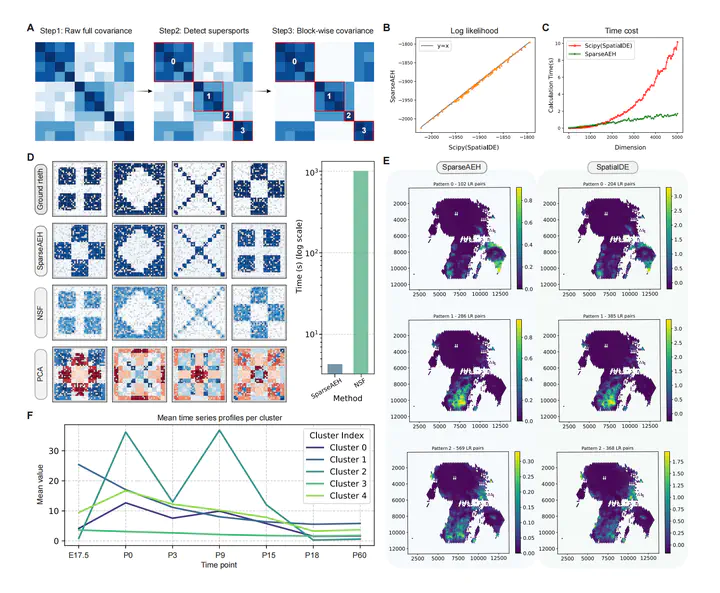 Image credit: [Lingyu Li]
Image credit: [Lingyu Li]
Abstract
Spatial clustering of genes is an effective way to elucidate histological or spatial patterns of marker expressions or ligand-receptor interactions. However, its computational complexity hinders its application to the advanced spatial transcriptomics (ST) technologies with high resolution and throughput. Here, we present SparseAEH, a highly scalable algorithm and implementation of the Gaussian process mixture model, by leveraging block-wise sparse covariance for approximating the likelihood. This acceleration enables the analysis of dense ST datasets within practical time constraints while maintaining accuracy. Benchmarking on both simulated and experimental data demonstrates that our method achieves significantly less computational time with comparable accuracy to previous methods, highlighting its potential as a powerful tool for the exploration of high-resolution ST data.
Supplementary notes can be added here, including code, data, math, and images.
Lingyu Li
Postdoctoral Fellow
Focus on bioinformatics, including but not limited to spatial transcriptomics analysis, sparse statistical learning and biomarker identification.