scGENA: A single-cell gene coexpression network analysis framework for clustering cell types and revealing biological mechanisms
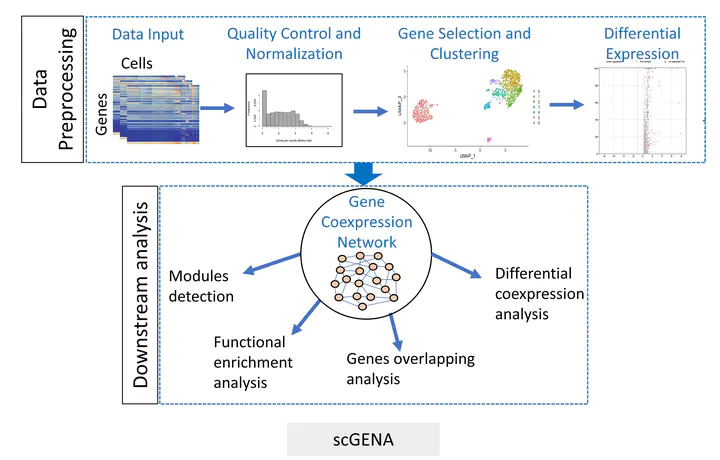 Image credit: Lingyu Li
Image credit: Lingyu Li
摘要
Single-cell RNA-sequencing (scRNA-seq) is a recent high-throughput technique that can measure gene expression, reveal cell heterogeneity, rare and complex cell populations, and discover cell types and their relationships. The analysis of scRNA-seq data is challenging because of transcripts sparsity, replication noise, and outlier cell populations. A gene coexpression network (GCN) analysis effectively deciphers phenotypic differences in specific states by describing gene–gene pairwise relationships. The underlying gene modules with different coexpression patterns partially bridge the gap between genotype and phenotype. This study presents a new framework called scGENA (single-cell gene coexpression network analysis) for GCN analysis based on scRNA-seq data. Although there are several methods for scRNA-seq data analysis, we aim to build an integrative pipeline for several purposes that cover primary data preprocessing, including data exploration, quality control, normalization, imputation, and dimensionality reduction of clustering as downstream of GCN analysis. To demonstrate this integrated workflow, an scRNA-seq dataset of the human diabetic pancreas with 1600 cells and 39,851 genes was implemented to perform all these processes in practice. As a result, scGENA is demonstrated to uncover interesting gene modules behind complex diseases, which reveal biological mechanisms. scGENA provides a state-of-the-art method for gene coexpression analysis for scRNA-seq data.
Supplementary notes can be added here, including code, data, math, and images.